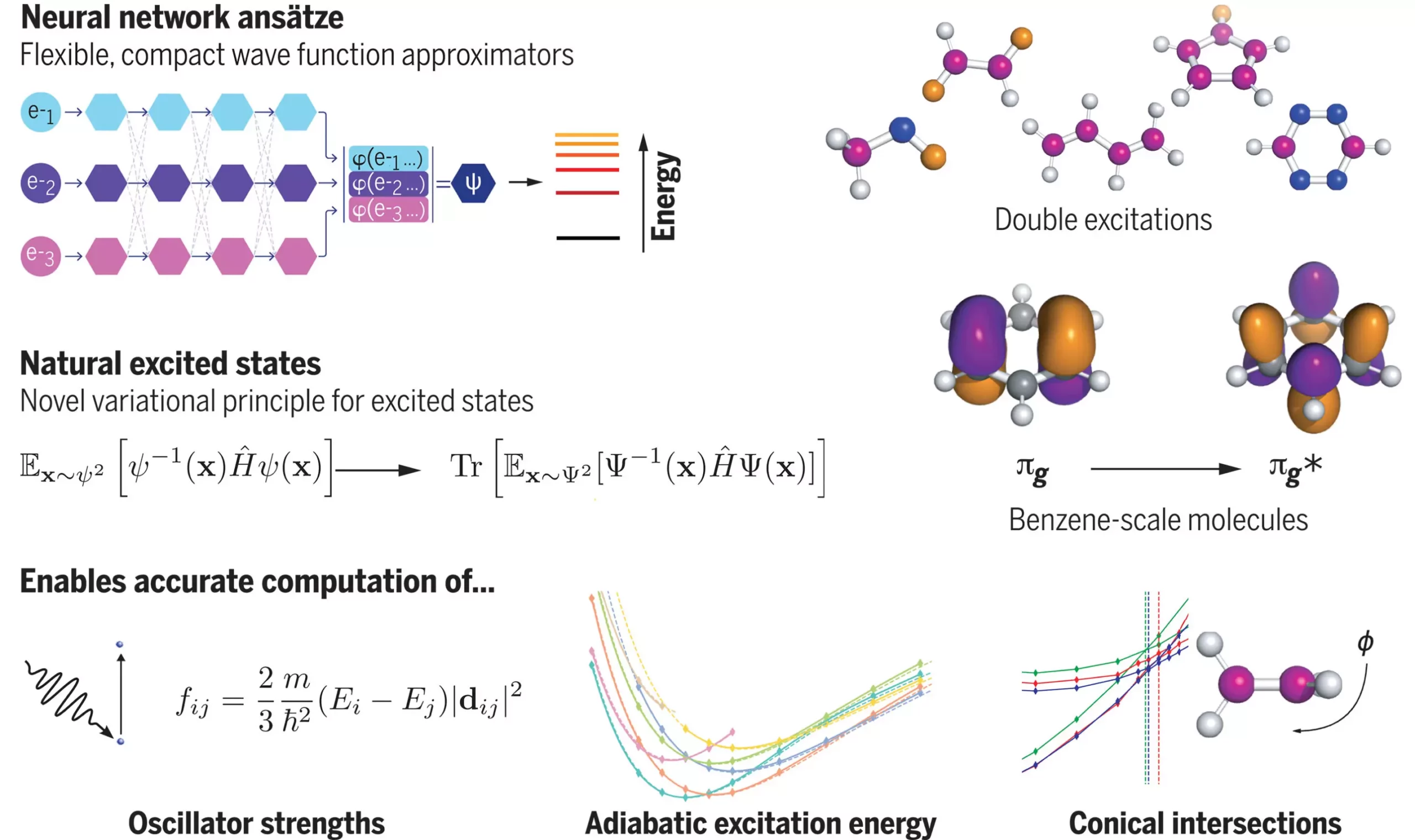
Recent research conducted by scientists from Imperial College London and Google DeepMind has unveiled a groundbreaking solution to the complex task of modeling the states of molecules. This innovative study harnesses the power of neural networks, a form of artificial intelligence inspired by the human brain, to tackle fundamental equations in intricate molecular systems. By delving into how molecules transition between different states, this research opens up exciting possibilities for the future of materials design and chemical synthesis through computer simulations.
When molecules and materials are exposed to high energy levels, such as light or heat, their electrons undergo transitions into temporary new configurations known as excited states. These transitions generate a unique energy fingerprint that varies for each molecule and material, influencing the performance of technologies like solar panels, LEDs, semiconductors, and photocatalysts. Moreover, these molecular transitions play a crucial role in biological processes involving light, such as photosynthesis and vision.
One of the main challenges in molecular modeling lies in accurately representing the quantum nature of excited electrons within molecules. These electrons do not have fixed positions but instead exist as probabilities across all possible configurations. The vast number of potential electron configurations poses a daunting task for traditional computational methods. Lead researcher Dr. David Pfau emphasizes the complexity of this task, stating that attempting to represent the electron configurations of a silicon atom would require a grid with a staggering number of points, exceeding the total number of atoms in the universe.
To address this computational challenge, the research team developed a novel mathematical approach and combined it with a neural network called FermiNet (Fermionic Neural Network). FermiNet represents a pioneering example of utilizing deep learning to compute the energy of atoms and molecules from first principles with remarkable accuracy. Through rigorous testing on various molecular examples, the team achieved promising results. Notably, when analyzing the carbon dimer molecule, they attained a mean absolute error of 4 meV, significantly closer to experimental results than previous methods with an error of 20 meV.
The success of this research opens up a myriad of possibilities for the field of computational chemistry and materials science. By accurately modeling molecular transitions and energy states, researchers can streamline the process of developing new materials and chemical syntheses through virtual simulations. This predictive capability not only accelerates the pace of scientific discovery but also reduces the costs and time associated with experimental trial and error in the lab. The utilization of neural networks in molecular modeling represents a paradigm shift in the way we approach complex quantum systems, offering a more efficient and accurate means of understanding and manipulating molecular behavior.
The integration of neural networks in molecular modeling holds immense promise for revolutionizing the way we study and design materials at the atomic level. By leveraging the power of artificial intelligence inspired by the human brain, researchers are pushing the boundaries of computational chemistry and paving the way for a new era of innovation in materials science.
Leave a Reply