At the heart of chemistry lies the atom—a minuscule entity made up of a positively charged nucleus, orbited by negatively charged electrons. The true intricacy of atomic behavior and its transformation into molecular dynamics makes simulating these interactions exceptionally challenging. As more atoms group to form a molecule, the number of interactions spikes, creating a computational conundrum. Conventional methods, heavily reliant on solving the Schrödinger equation, struggle to keep pace with the demands of modern science. This equation, which delineates the energy levels of quantum systems, becomes computationally overwhelming even for relatively small molecules. Traditional techniques may take days to compute, especially when grappling with molecules more complex than a few dozen atoms. This protracted timeline not only hampers research efforts but also inflates costs and environmental implications tied to extensive computational resources.
Machine Learning as a Game Changer
Recent advances, particularly within machine learning (ML), are beginning to shift this paradigm. Researchers from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) and Google DeepMind have innovated a new learning algorithm that enables surprisingly accurate simulations of molecular dynamics over lengthy time scales. This breakthrough is especially crucial for applications in drug design and the development of materials, such as those utilized in solar panels or batteries. By leveraging the predictive power of ML, scientists are now able to circumvent the arduous task of solving the Schrödinger equation directly. Instead, they focus on teaching machines to predict electronic interactions at an atomic level.
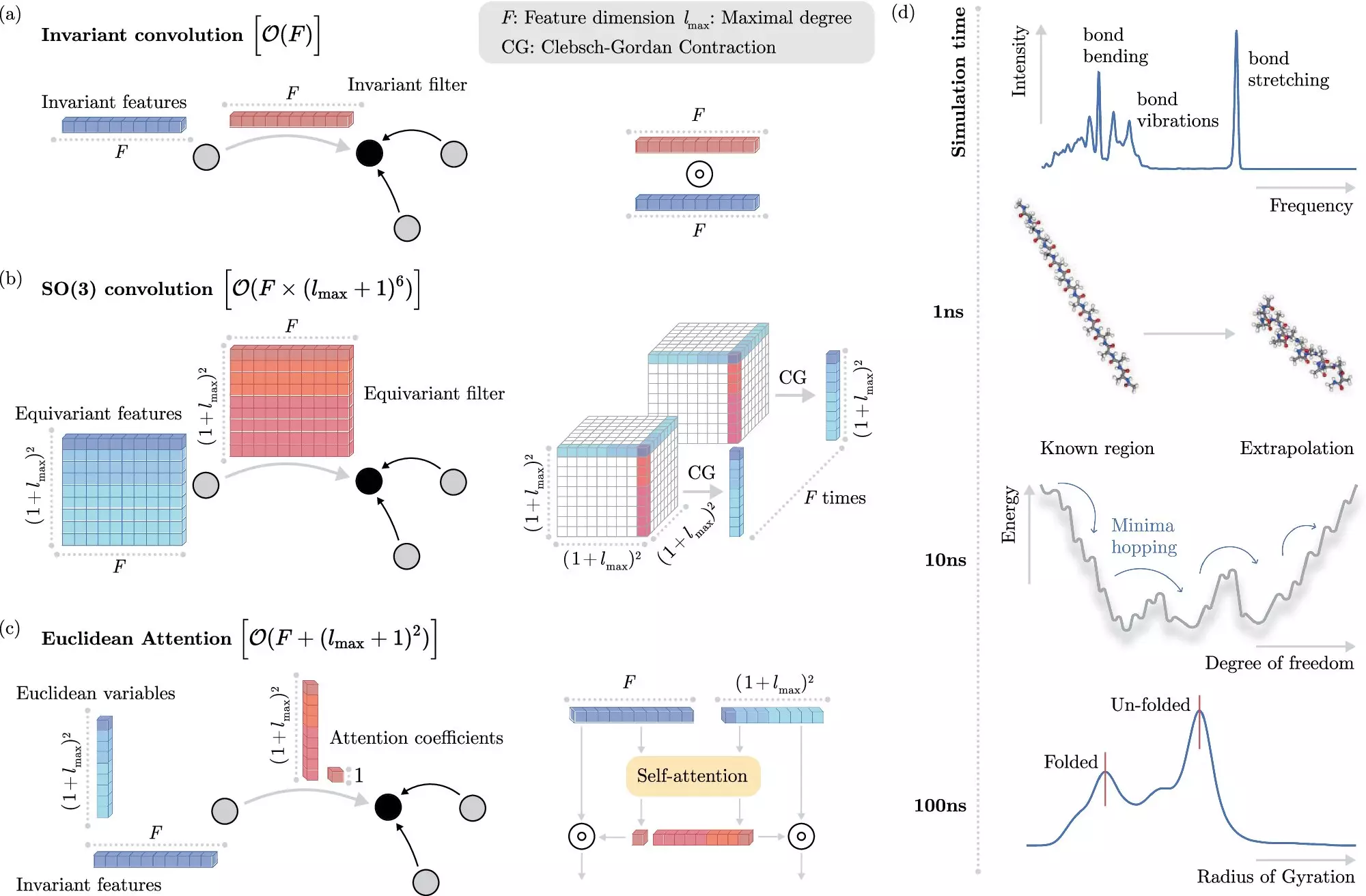
However, the complexity of teaching these systems is not minor. It requires a sophisticated understanding of how atomic interactions manifest in real-world scenarios. The challenge lies in revealing how electrons interact without attempted simulations or explicit modeling of every variable. The emergence of algorithms that recognize “invariances” within physical systems offers a beacon of hope; certain properties remain unchanged as molecules shift in space, allowing the ML models to generalize knowledge rather than learn from scratch every time.
New Paradigms in Algorithm Design
The novel algorithm developed by the BIFOLD team significantly enhances the efficiency of molecular simulations. This work involves decoupling invariances from other components of chemical systems right from the beginning. Traditional methods often required the tedious extraction of invariant aspects during each modeling step, thus limiting computational speed and feasibility for long-term simulations. However, the new ML approach simplifies this procedure, allowing researchers to focus on significant interactions that contribute to molecular behavior while reducing overall computational expenses.
Dr. Stefan Chmiela, who spearheaded this research, notes that the time required for simulations has drastically reduced from months or even years down to just a few days on standard computer nodes. This efficiency leap is not just a minor improvement; it empowers researchers to simulate interactions over extended timeframes necessary for a deeper understanding of atomic systems. The implications here are staggering; insights that were once confined to extensive theoretical frameworks and expensive physical experiments are now within reach, essentially bridging the gap between molecular dynamics and real-time computational analysis.
Applications: The Future of Drug Development
Consider the potential ramifications of this research for the pharmaceutical industry. In a health landscape that increasingly prioritizes quick and cost-effective drug development, the possibility of accurately simulating how molecules interact with proteins in the human body paves the way for revolutionary advancements. By employing this new algorithm, researchers can explore possible drug candidates without the need for extensive physical experimentation, thus converting previously impractical research timelines into actionable insights.
One example of this application includes the identification of the stable structure of docosahexaenoic acid, a critical component of the human brain. With the capacity to assess tens of thousands of configurations with high precision, scientists can bypass traditional quantum mechanical limitations that had rendered such tasks unfeasible until now. As echoed by Prof. Dr. Klaus-Robert Müller, co-director of BIFOLD, the capacity to integrate cutting-edge machine learning with fundamental physical principles could surmount long-standing hurdles in computational chemistry.
A Bright Horizon for Computational Chemistry
As algorithms evolve, the future demands an accurate representation of complex physical interactions, particularly long-range ones. The journey towards achieving scalable ML approaches for real-world chemical systems continues to be an exhilarating frontier in science. The amalgamation of machine learning with the foundational aspects of quantum mechanics opens doors not just for understanding but for practical applications that were previously inconceivable. In unveiling the mysteries of molecular dynamics, we are on the cusp of a scientific revolution that promises to reshape fields from healthcare to energy solutions. The future, illuminated by innovation, beckons researchers to explore untested waters, potentially leading to breakthroughs that could have unprecedented impacts.
Leave a Reply